lunes, 31 de octubre de 2016
domingo, 30 de octubre de 2016
sábado, 29 de octubre de 2016
jueves, 27 de octubre de 2016
Electrophoresis en gel de Poliacrilamida
Electrophoresis de proteinas en gel de poliacrilamida

http://www.ub.es/biocel/wbc/tecnicas/page.htm#Electroforesis de proteínas en geles de poliacrilamida

La electroforesis de proteínas en geles con una matriz de poliacrilamida, comunmente denominada electroforesis en poliacrilamida (PAGE, 'polyacrilamide gel electrophoresis') es sin duda alguna una de las técnicas más ampliamente usada para caracterizar mezclas complejas de proteínas. La electroforesis en poliacrilamida es un método conveniente, rápido y económico a nivel de muestra pues se requieren sólo cantidades del orden de microgramos de proteína.
Las proteínas presentan una carga eléctrica neta si se encuentran en un medio que tenga un pH diferente al de su punto isoeléctrico y por eso tienen la propiedad de desplazarse cuando se someten a un campo eléctrico. La velocidad de migración es proporcional a la relación entre las cargas de la proteína y su masa. Cuanto mayor carga por unidad de masa más rápida será la migración. Empleando geles de sílice o de acetato de celulosa y aplicando las proteínas en una zona estrecha en torno a los electrodos se pueden determinar diferencias de carga neta (carga total/masa) entre proteínas. Este método se denomina electroforesis zonal. La matriz de poliacrilamida no es un buen soporte para este método pues la migración de las proteínas en su seno no sólo es proporcional a la carga neta sino también al tamaño y forma de las proteínas..
Una ventaja importante de los geles de poliacrilamida es que son químicamente inertes, transparentes y estables en un amplio rango de pHs, temperatura y fuerza iónica.
http://www.ub.es/biocel/wbc/tecnicas/page.htm#Electroforesis de proteínas en geles de poliacrilamida
sábado, 8 de octubre de 2016
Purificación de Proteínas

Las separaciones cromatográficas se basan en las diferencias de carga, tamaño o afinidad de las diferentes proteínas. Uno de los métodos más habitualmente utilizados para la separación de proteínas es la cromatografía en columna, aunque también existe la cromatografía en papel y en placa.
La columna está rellena con un material sólido con las características químicas adecuadas (fase estacionaria), y una solución tamponada se hace pasar a través de la columna (fase móvil). El extracto crudo se aplica en la parte superior de la columna y va poco a poco entrando en la columna con la fase móvil. Cada proteína migrará a distinta velocidad según su grado de afinidad por la fase estacionaria.
Generalmente estas cromatografías se realizan de una forma semiautomática, la fase móvil se hace pasar a la columna a través de una bomba peristáltica y un colector de fracciones va recogiendo el eluyente que sale de columna. Este colector hace que el tubo vaya cambiando cada cierto número de gotas o de volumen. Después hay que localizar en que tubo o tubos está la proteína que se pretende purificar.
Existen distintos tipos de cromatografías algunos de ellos son:
Filtración en gel o cromatografía de
exclusión molecular. La fase estacionaria esta
constituida por partículas de polímeros de
diferente porosidad. La separación se basa en el
tamaño de las partículas. Las proteínas más
grandes que no pueden penetrar en los poros de
las partículas de la matriz de filtración son
eluidas con más rapidez que las proteínas más
pequeñas que penetran por los poros de las
partículas y siguen un camino más tortuoso y
más largo. El tamaño de los poros internos
depende de la naturaleza del polímero en
cuestión, y permite la entrada a proteínas por
debajo de un determinado peso molecular.
Cromatografía de intercambio iónico: En ella, la columna está rellena con un soporte al que van unidos grupos cargados positivamente (intercambio aniónico) o negativamente (intercambio catiónico). Estos grupos cargados normalmente están neutralizados por iones del tampón. Estos iones son reversiblemente reemplazados por las proteínas con más tendencia a unirse al soporte. Las proteínas cargadas pueden por lo tanto unirse a intercambiadores catiónicos o aniónicos dependiendo de su carga neta. Las proteínas más
cargadas se unirán más fuertemente al intercambiador y por lo tanto serán más difíciles de eluir. La afinidad con la que una proteína se une a un intercambiador iónico depende de la fuerza iónica del medio, debido a la competencia entre los grupos cargados de la proteína y los iones de la fase móvil.
Una vez pegadas las proteínas, para eluirlas (retirarlas) de la columna, se suele ir subiendo la fuerza iónica de la fase móvil (aumentando la concentración de sal, NaCl), de esta forma se eluyen primero las proteínas mas débilmente retenidas y cuando la fuerza iónica sea mayor saldrán las proteínas más cargadas y por lo tanto más retenidas.
Cromatografía hidrofóbica: Se trata de un método similar al anterior con la única diferencia de que la fase estacionaria es apolar (hidrofóbica), es decir, la proteína se pega al soporte por los resto de aminoácidos apolares, cuanto más residuos de este tipo tenga en su superficie, más apolar será, más se retendrá en la columna y se eluirá más tarde. En este caso a elusión se realiza aumentando la apolaridad de fase móvil.
Cromatografía de afinidad: Muchas proteínas tienen la capacidad de unirse específicamente a ciertas moléculas, mediante uniones fuertes, pero no covalentes. En esta técnica la molécula que se une específicamente a la proteína se conoce con el nombre de ligando y se une covalentemente a una matriz inerte. Cuando el extracto con la mezcla de proteínas se aplica a la columna, la proteína buscada se unirá al ligando inmovilizado mientras que las demás saldrán de la columna con el tampón. En este caso también se utiliza el incremento de fuerza iónica para eluir las proteínas.
Cromatografía de intercambio iónico: En ella, la columna está rellena con un soporte al que van unidos grupos cargados positivamente (intercambio aniónico) o negativamente (intercambio catiónico). Estos grupos cargados normalmente están neutralizados por iones del tampón. Estos iones son reversiblemente reemplazados por las proteínas con más tendencia a unirse al soporte. Las proteínas cargadas pueden por lo tanto unirse a intercambiadores catiónicos o aniónicos dependiendo de su carga neta. Las proteínas más
cargadas se unirán más fuertemente al intercambiador y por lo tanto serán más difíciles de eluir. La afinidad con la que una proteína se une a un intercambiador iónico depende de la fuerza iónica del medio, debido a la competencia entre los grupos cargados de la proteína y los iones de la fase móvil.
Una vez pegadas las proteínas, para eluirlas (retirarlas) de la columna, se suele ir subiendo la fuerza iónica de la fase móvil (aumentando la concentración de sal, NaCl), de esta forma se eluyen primero las proteínas mas débilmente retenidas y cuando la fuerza iónica sea mayor saldrán las proteínas más cargadas y por lo tanto más retenidas.
Cromatografía hidrofóbica: Se trata de un método similar al anterior con la única diferencia de que la fase estacionaria es apolar (hidrofóbica), es decir, la proteína se pega al soporte por los resto de aminoácidos apolares, cuanto más residuos de este tipo tenga en su superficie, más apolar será, más se retendrá en la columna y se eluirá más tarde. En este caso a elusión se realiza aumentando la apolaridad de fase móvil.
Cromatografía de afinidad: Muchas proteínas tienen la capacidad de unirse específicamente a ciertas moléculas, mediante uniones fuertes, pero no covalentes. En esta técnica la molécula que se une específicamente a la proteína se conoce con el nombre de ligando y se une covalentemente a una matriz inerte. Cuando el extracto con la mezcla de proteínas se aplica a la columna, la proteína buscada se unirá al ligando inmovilizado mientras que las demás saldrán de la columna con el tampón. En este caso también se utiliza el incremento de fuerza iónica para eluir las proteínas.
sábado, 1 de octubre de 2016
lunes, 26 de septiembre de 2016
Transformación bacteriana
Transformación Bacteriana
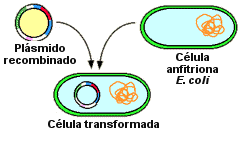
Transformación
bacteriana es un procedimiento de laboratorio por
el cual se introduce material genético a una bacteria. Existen
otros procesos de inserción del material genético, que dependiendo del
organismo receptor y el mecanismo, algunos reciben nombres como transfección o traducción.
Generalmente, el material genético insertado es conocido
como plasmado (DNA circular), pero pueden insertarse otras formas de
material genético, como DNA o RNA. Los plásmidos utilizados
para la transformación de bacterias contienen en general, uno o varios genes de
interés, un gen reportero (que permite visualizar la transcripción del
plásmido), y un gen de resistencia a un antibiótico, que permite seleccionar a
la bacteria transformada de otras, por su capacidad de crecer en medio que
contiene el antibiótico de resistencia.
Las transformación tienen muchas aplicaciones, como la
producción de proteínas, la producción de los mismos plásmidos, la producción
de bacteria que consumen petróleo, entre otros. Gen es un
pedazo de DNA el cual provee las instrucciones para hacer (o codifica para) una
proteína. Esta proteína le da a un organismo una característica particular. La
transformación genética ocurre cuando una célula incorpora dentro de ella y
expresa un nuevo pedazo de material genético ( DNA). Esta nueva
información genética muchas veces provee al organismo de una nueva
característica que se puede identificar luego de ocurrida la
transformación Transformación genética significa cambio causado por
genes e involucra la inserción de uno o más genes en el organismo en orden de
cambiar las características del organismo.
Vídeo de lo que es transformación bacteriana
Vídeo de lo que es transformación bacteriana
Micropipetas: Descripción general

MICROPIPETAS
Descripción del equipo
La micropipeta es un instrumento de laboratorio empleado para absorber y transferir pequeños volúmenes de líquidos y permitir su manejo en las distintas técnicas científicas.
Los volúmenes captables por estos instrumentos varían según el modelo: los más habituales, denominados p20, p200 y p1000, admiten un máximo de 20, 200 y 1000 μl, respectivamente. Es de destacar que el uso de micropipetas permite emplear distintos líquidos sin tener que lavar el aparato: para ello, se emplean puntas desechables, de plástico, que habitualmente son estériles.
La micropipeta es un instrumento de laboratorio empleado para absorber y transferir pequeños volúmenes de líquidos y permitir su manejo en las distintas técnicas científicas.
Los volúmenes captables por estos instrumentos varían según el modelo: los más habituales, denominados p20, p200 y p1000, admiten un máximo de 20, 200 y 1000 μl, respectivamente. Es de destacar que el uso de micropipetas permite emplear distintos líquidos sin tener que lavar el aparato: para ello, se emplean puntas desechables, de plástico, que habitualmente son estériles.
Operación del Equipo
Técnica de pipeteo para líquidos claros:
a. Se presiona el botón superior suavemente hasta el primer tope.
b. Se sumerge la punta, en la solución que se necesita pipetear estando
seguros que la punta este bien colocada y que no haya ningún tipo de
residuos entre la punta y el cuerpo de la pipeta.
c. Mantenga la pipeta verticalmente mientras toma la solución.
d. Para descartar la solución de la punta presione el botón hasta el segundo
tope.
e. Descarte las puntas utilizando el eyector que traen las pipetas.
Técnica de pipeteo para líquidos claros:
a. Se presiona el botón superior suavemente hasta el primer tope.
b. Se sumerge la punta, en la solución que se necesita pipetear estando
seguros que la punta este bien colocada y que no haya ningún tipo de
residuos entre la punta y el cuerpo de la pipeta.
c. Mantenga la pipeta verticalmente mientras toma la solución.
d. Para descartar la solución de la punta presione el botón hasta el segundo
tope.
e. Descarte las puntas utilizando el eyector que traen las pipetas.
viernes, 2 de septiembre de 2016
Electrophoresis en gel de agarosa

Electroforesis en Gel de Agarosa
La electroforesis es una técnica habitual en el laboratorio clínico o de investigación. Permite separar especies químicas (ácidos nucleicos o proteínas) a lo largo de un campo eléctrico en función de su tamaño y de su carga eléctrica. Los ácidos nucleicos, ADN y ARN, tienen por naturaleza carga negativa. Si ponemos fragmentos del ADN extraído de una muestra biológica sobre un soporte poroso (gel) y aplicamos un campo eléctrico, se producirá la migración diferencial de los fragmentos a través de los poros de la matriz. La tinción del gel con bromuro de etidio, que es una sustancia fluorescente que se intercala en la molécula de ADN, y la exposición con luz ultravioleta, permite observar el resultado de ésta migración. El tamaño de los fragmentos de ADN se puede estimar comparándolos con el patrón de bandas que se obtiene de marcadores comerciales de peso molecular conocido. La electroforesis de ADN es útil para comparar patrones de bandas de diferentes muestras biológicas. Así por ejemplo se puede usar para comparar muestras de individuo sano frente a individuo enfermo, para la identificación de ácidos nucleicos de agentes infecciosos o para la obtención de un fragmento determinado de ADN que, una vez localizado en el gel, puede ser extraído para análisis posteriores.
lunes, 29 de agosto de 2016
Reacción en Cadena de la Polimerasa (PCR)

Es una reacción enzimática in vitro que amplifica millones de veces una secuencia específica de ADN durante varios ciclos repetidos en los que la secuencia blanco es copiada fielmente. Para ello, la reacción aprovecha la actividad de la enzima ADN polimerasa que tiene la capacidad de sintetizar naturalmente el ADN en las células. En la reacción, si usamos como sustrato ADN genómico, entonces típicamente hablamos de una PCR, pero si usamos ADN complementario (ADNc) proveniente del ARNm (ácido ribonucleico mensajero) se le conoce como RT-PCR (Reverse Transcription-PCR, por sus siglas en inglés). Esta conversión se logra mediante una reacción conocida como transcripción reversa y controlada por la enzima transcriptasa reversa, capaz de convertir el ARNm en una molécula de ADNc.
Cada ciclo de la PCR se lleva a
cabo en tres etapas principales: desnaturalización,
hibridación y extensión:
Desnaturalización. En esta etapa, las cadenas de
ADN son calentadas y separadas a una temperatura de
95 °C durante 20-30 segundos; el tiempo depende de
la secuencia del templado, es decir, si la cantidad de
G-C es alta, será necesario más tiempo para romper
sus uniones debido a que el apareamiento de estas
bases está formado por tres enlaces, uno más que las
bases de A-T. Además, depende de la velocidad en
la que el termociclador aumenta la temperatura, esto
varía de acuerdo al modelo del equipo. Al final de esta
etapa tendremos las cadenas separadas que servirán
como templado para el siguiente paso.
Hibridación. En esta etapa, los primers se alinean al extremo 3’ del templado previamente separado e hibridan con su secuencia complementaria. Para que se forme el complejo templado-primers, es importante que la temperatura de hibridación o temperatura melting (Tm) sea la óptima; ésta generalmente oscila entre 50-60 °C. Si el diseño de los primers es el correcto y la temperatura es la adecuada, la estabilidad y especificidad del complejo será eficiente.
Extensión. En esta etapa, la Taq polimerasa actúa sobre el complejo templado-primers y empieza su función catalítica a una velocidad muy rápida; agrega dNTP’s complementarios para crear las cadenas completas de ADN. La extensión de las cadenas es en dirección de la síntesis del ADN, es decir, de 5’ a 3’. La temperatura óptima para la reacción es de 72 °C, ya que a esa temperatura la enzima es funcional. Al final del ciclo, se habrán formado los amplicones con un tamaño dictado por el número total de pares de bases (pb) que deberá ser conocido por el investigador.
Mitos y Realidades:
http://www.microbial-systems.com/web/docs/Newsletter_Microbial_03.pdf
Hibridación. En esta etapa, los primers se alinean al extremo 3’ del templado previamente separado e hibridan con su secuencia complementaria. Para que se forme el complejo templado-primers, es importante que la temperatura de hibridación o temperatura melting (Tm) sea la óptima; ésta generalmente oscila entre 50-60 °C. Si el diseño de los primers es el correcto y la temperatura es la adecuada, la estabilidad y especificidad del complejo será eficiente.
Extensión. En esta etapa, la Taq polimerasa actúa sobre el complejo templado-primers y empieza su función catalítica a una velocidad muy rápida; agrega dNTP’s complementarios para crear las cadenas completas de ADN. La extensión de las cadenas es en dirección de la síntesis del ADN, es decir, de 5’ a 3’. La temperatura óptima para la reacción es de 72 °C, ya que a esa temperatura la enzima es funcional. Al final del ciclo, se habrán formado los amplicones con un tamaño dictado por el número total de pares de bases (pb) que deberá ser conocido por el investigador.
Mitos y Realidades:
http://www.microbial-systems.com/web/docs/Newsletter_Microbial_03.pdf
Extracción de DNA

Extracción de Ácido Desoxirribonucleico (DNA):
La extracción de DNA consta de una etapa de lisis, que consiste en romper las estructuras que confinan el citoplasma y liberar al medio su contenido y otra de purificación, que implica la retirada de la solución final de la mayoría de elementos que pueden interferir en la PCR.
Etapas en la extracción de DNA
Los pasos necesarios para una correcta extracción y purificación del DNA mediante un proce- dimiento químico son:
-
Lisis de las células o virus. Las sales caotrópicas ayudan a romper la estructura tridimensional de macromoléculas como las
proteínas o los ácidos nucleicos consiguiendo su desnaturalización. La adición de un
detergente como el SDS es necesaria a menudo para eliminar las membranas.
-
Degradación de la fracción proteica asociada al DNA. Se consigue mediante la adición
de una proteasa. La fracción proteica puede
precipitarse mejor con la ayuda de sales
como el acetato de amonio o el acetato
sódico.
- Purificación. Consta de 3 fases. Precipitación del DNA. El DNA es insoluble en alcohol, por lo que se puede precipitar etanol frío o isopropanol i recuperar mediante una centrifugación. El alcohol del sobrenadante se llevará las sales añadidas previamente.
Célula Eucariota Animal

La célula eucariota animal presenta las siguientes estructuras:
1. Membrana plasmática
2. Citoplasma
3. Núcleo
4. Orgánulos:
− Sin membrana
- Ribosomas: Pequeños orgánulos celulares, constituidos por ARN y proteínas encargados de la síntesis de proteínas.
- Citoesqueleto: Conjunto de filamentos proteicos que forman redes complejas. Mantienen la forma celular e intervienen en el movimiento de orgánulos y división celular.
- Centriolos: Cilindros formados por túbulos que dirigen el movimiento de cilios y flagelos y participan en el reparto del material genético durante la división celular.
Célula Eucariota
Célula eucariota:
La célula eucariota se caracteriza por la presencia de un núcleo en cuyo interior se encuentra el material genético. Las células eucariotas son mayores que las procariotas; tienen un tamaño que oscila entre las 10 y las 100 µm.
A pesar de que existen dos tipos de células eucariotas (animal y vegetal) tienen en común las siguientes estructuras:
- Membrana plasmática: Es una envoltura o capa formada principalmente por lípidos y proteínas. Delimita la célula, le da forma y permite el intercambio de sustancias con el medio externo.
- Citoplasma: Es el espacio comprendido entre la membrana plasmática y el núcleo. Está formado por una sustancia líquida y viscosa en la que se encuentran inmersos los orgánulos, es decir, los diversos componentes de la célula encargados de diferentes funciones.
- Núcleo: Se encuentra en el interior de la célula, posee una doble membrana en cuyo interior se encuentra el material genético.
La célula eucariota se caracteriza por la presencia de un núcleo en cuyo interior se encuentra el material genético. Las células eucariotas son mayores que las procariotas; tienen un tamaño que oscila entre las 10 y las 100 µm.
A pesar de que existen dos tipos de células eucariotas (animal y vegetal) tienen en común las siguientes estructuras:
- Membrana plasmática: Es una envoltura o capa formada principalmente por lípidos y proteínas. Delimita la célula, le da forma y permite el intercambio de sustancias con el medio externo.
- Citoplasma: Es el espacio comprendido entre la membrana plasmática y el núcleo. Está formado por una sustancia líquida y viscosa en la que se encuentran inmersos los orgánulos, es decir, los diversos componentes de la célula encargados de diferentes funciones.
- Núcleo: Se encuentra en el interior de la célula, posee una doble membrana en cuyo interior se encuentra el material genético.
Célula Procariota

Célula procariota
Las células procariotas se caracterizan por no
presentar núcleo. Son células de menor tamaño que las eucariotas y suelen medir
entre 1 y 10 µm. Los seres vivos más representativos de este tipo son las
bacterias.
Presentan las siguientes
estructuras:
1. Membrana
plasmática: Estructura flexible que delimita la célula y permite y regula
los intercambios con el medio exterior. Tiene unos repliegues internos, los
donde tiene lugar la mayoría de las reacciones químicas propias de la nutrición
celular.
2. Pared
bacteriana: Envoltura rígida, externa a la membrana plasmática.
3. Citoplasma:
Espacio que ocupa el interior de la célula, constituido por una sustancia
líquida y otros componentes celulares inmersos en él.
4. Ribosomas:
Orgánulos encargados de la síntesis de proteínas. Son los únicos orgánulos
presentes en las células procariotas.
5. Material
genético: Cadena de ADN que flota libremente en el citoplasma.
6. Flagelo:
Lo presenta algunos grupos de bacterias. Los flagelos son estructuras
filamentosas que salen al exterior desde la membrana plasmática y permiten el
movimiento de la célula.
La forma de las células bacterianas es muy
diversa, algunas de ellas son:
− Bacilos: Forma de
bastón
− Cocos: Forma
esférica
− Espirilos: Forma de
tirabuzón
− Vibrios: Forma de coma.
Estructura celular
Todas las células presentan una estructura común
formada por: membrana plasmática, citoplasma y material genético.
Existen dos tipos de estructura celular:
• Célula procariota
• Célula eucariota
−
Animal
−Vegetal
Suscribirse a:
Entradas (Atom)